Welcome to MergeMapMergeMap is a software tool that is capable of constructing accurate consensus genetic maps from a set of individual genetic maps. The input to MergeMap, a set of individual maps, are first converted to DAGs internally, which are then merged into a consensus graph on the basis of shared vertices. Conflicts among the individual maps will be shown as cycles in the consensus graph. MergeMap tries to resovle conflicts by deleting a minimum set of marker occurrences. The details about the conflicts as well the decision by MergeMap are shown to the user graphically. The result of this conflict-resolution step is a consensus DAG, which will be simplified and then linearized to produce the final consensus map. The consensus map is in the same format as the input genetic maps. For details of our algorithm, please refer to our paper titled "On the Accurate Construction of Consensus Genetic Maps", which is to appear in CSB 2008. The format of the individual genetic linkage mapsThe individual genetic linkage map is in the following format:
group lg1
;BEGINOFGROUP
m0 0.000
m8 11.183
m7 11.183
m6 12.183
......
m94 175.020
m95 179.032
m97 183.083
m98 183.083
;ENDOFGROUP
.
.
.
group lg2
;BEGINOFGROUP
m100 0.000
m108 11.183
m107 11.183
m106 12.183
......
m194 175.020
m195 179.032
m197 183.083
m198 183.083
;ENDOFGROUP
Each individual genetic map may consists of multiple linkage groups (LG).
Each LG starts with the line "group group_name",
where group_name is the name for that LG, followed by the line ";BEGINOFGROUP", and
then the main body of the LG. It ends with the line ";ENDOFGROUP" indicating the end of the LG.
The main body of the LG consists of multiple lines, each of which corresponds to one marker. Each line
specifies the name of the marker as well as the distance (in cm) from the marker to the first marker
in the LG.
How to use MergeMapYou need to first download the source for MergeMap and then compile it on a linux machine. MergeMap depends on the boost library; therefore you will also need to download and install the boost library if you don't have it on your linux machine, and then edit the Makefile to correctly point it to the directory where the boost library resides. To use MergeMap, you will need to first construct a configuration file in the following format:
map1_name map1_weight map1_path
map2_name map2_weight map2_path
map3_name map3_weight map3_path
...
Each line of the configuration file refers to one individual genetic linkage map.
It consists of three entries separated by blank spaces.
The first entry specifies the name of the linkage map; the second entry specifies
the weight of the linkage map; and the last entry specifies the path to the individual map.
The weight represents the user's confidence in the quality of the map. The more confidence
one has, the higher the weight should be. When MergeMap tries to resolve conflicts,
it will preferably delete marker occurrences from the map of the lowest weight.
Once the input genetic maps and the configuration file are ready, one can simply run the following command to construct the consensus map:
MergeMap configuration_file
The format of the output filesThe LGs from the individual genetic maps are first divided into clusters according to their marker composition. Two LGs belong to the same cluster if they share any markers in common. Each cluster corresponds to a linkage group in the consensus map. MergeMap then processes the clusters sequentially. For each cluster, MergeMap first identifies a consistent orientation by flipping some of the constituent LGs. It then produces a consensus DAG of the cluster by resolving the conflicts (if there is any). The consensus DAG is further simplified and then linearized to give the final consensus map. For each cluster, three graphs in the .dot format are produced. They are saved as lgx.dot, lgx_consensus.dot, and lgx_linear.dot files respectively, where x is the id of the cluster. These graphs can be visualized with the GraphViz software tool, which is freely available at http://www.graphviz.org/. The lgx.dot graph highlights the conflicts among the individual maps. It also shows the solution by MergeMap as to which marker occurrence is being deleted. An example along with a detailed explanation is given in the following figure. The lgx_consensus.dot shows the simplified consensus DAG while the lgx_linear.dot shows the final linearied consensus map. click on the figure to zoom in
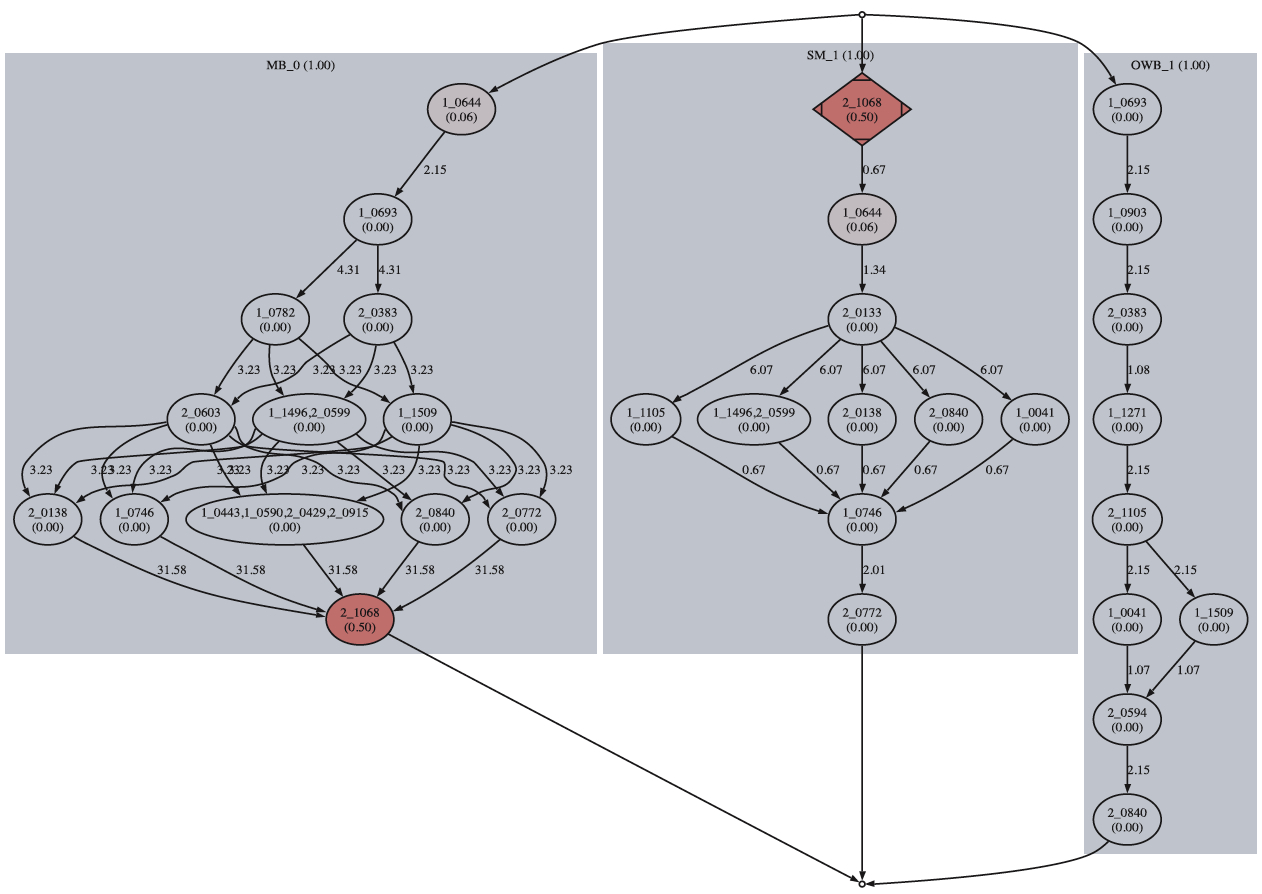 A fragment of the lgx.dot graph produced by MergeMap. This graph highlights the conflicts
among the three maps, namely the OWB map, the SM map and the MB map when building the
consensus map for chromosome 1H of barley. In the above figure each individual map is
represented as a shaded block. Marker ids are all of the form d_dddd where d is a digit.
The numbers on the edges indicate the distances between adjacent bins. The markers at
the same horizontal level belong to the same bin. The numbers enclosed in the parentheses
are the probabilities for deletion associated with the corresponding markers occurrences.
Intuitively this probability reflects how likely the marker occurrence is the trouble maker
that should be removed from the individual map. Each node is
filled with a color whose saturation is proportional to the associated probability (the hue
and brightness are constants). The higher the probability is,
the more standing out the color will be. This allows the end user to quickly spot the problematic markers.
The marker occurrences deleted by MergeMap are those enclosed in diamonds.
A fragment of the lgx.dot graph produced by MergeMap. This graph highlights the conflicts
among the three maps, namely the OWB map, the SM map and the MB map when building the
consensus map for chromosome 1H of barley. In the above figure each individual map is
represented as a shaded block. Marker ids are all of the form d_dddd where d is a digit.
The numbers on the edges indicate the distances between adjacent bins. The markers at
the same horizontal level belong to the same bin. The numbers enclosed in the parentheses
are the probabilities for deletion associated with the corresponding markers occurrences.
Intuitively this probability reflects how likely the marker occurrence is the trouble maker
that should be removed from the individual map. Each node is
filled with a color whose saturation is proportional to the associated probability (the hue
and brightness are constants). The higher the probability is,
the more standing out the color will be. This allows the end user to quickly spot the problematic markers.
The marker occurrences deleted by MergeMap are those enclosed in diamonds.
DownloadsSample Input is a synthetic data set with 100 markers and 8 individual maps. The true order of the markers is m0,m1,m2,...,m99 (or its reverse).CopyrightMergeMap is free for academic use only. For questions about the tool, please contact yonghui@cs.ucr.edu. |